Find and Highlight the Maximum Common Substructure Between a Set of Molecules Using RDKit
When analyzing a set of molecules, you might want to find the maximum common substructure (MCS) match between them. This utility function SmilesMCStoGridImage does that for a set of molecules specified by SMILES, displays the SMARTS substructure as a molecule, and displays all the molecules in a grid with that substructure highlighted and aligned.
Download this notebook from GitHub by right-clicking and choosing Save Link As…
The key RDKit commands it uses are:
FindMCSto find the maximum common substructure (SMARTS string)MolFromSmartsto generate a molecule corresponding to the maximum common substructureGenerateDepictionMatching2DStructureto align the matching substructureMolsToGridImageto draw the grid of the MCS, and the molecules with that MCS highlighted
from rdkit.Chem import AllChem as Chem
from rdkit.Chem import Draw
from rdkit.Chem import rdFMCS
def SmilesMCStoGridImage(smiles: list[str] or dict[str, str], align_substructure: bool = True, verbose: bool = False, **kwargs):
"""
Convert a list (or dictionary) of SMILES strings to an RDKit grid image of the maximum common substructure (MCS) match between them
:returns: RDKit grid image, and (if verbose=True) MCS SMARTS string and molecule, and list of molecules for input SMILES strings
:rtype: RDKit grid image, and (if verbose=True) string, molecule, and list of molecules
:param molecules: The SMARTS molecules to be compared and drawn
:type molecules: List of (SMARTS) strings, or dictionary of (SMARTS) string: (legend) string pairs
:param align_substructure: Whether to align the MCS substructures when plotting the molecules; default is True
:type align_substructure: boolean
:param verbose: Whether to return verbose output (MCS SMARTS string and molecule, and list of molecules for input SMILES strings); default is False so calling this function will present a grid image automatically
:type verbose: boolean
"""
mols = [Chem.MolFromSmiles(smile) for smile in smiles]
res = rdFMCS.FindMCS(mols, **kwargs)
mcs_smarts = res.smartsString
mcs_mol = Chem.MolFromSmarts(res.smartsString)
smarts = res.smartsString
smart_mol = Chem.MolFromSmarts(smarts)
smarts_and_mols = [smart_mol] + mols
smarts_legend = "Max. substructure match"
# If user supplies a dictionary, use the values as legend entries for molecules
if isinstance(smiles, dict):
mol_legends = [smiles[molecule] for molecule in smiles]
else:
mol_legends = ["" for mol in mols]
legends = [smarts_legend] + mol_legends
matches = [""] + [mol.GetSubstructMatch(mcs_mol) for mol in mols]
subms = [x for x in smarts_and_mols if x.HasSubstructMatch(mcs_mol)]
Chem.Compute2DCoords(mcs_mol)
if align_substructure:
for m in subms:
_ = Chem.GenerateDepictionMatching2DStructure(m, mcs_mol)
drawing = Draw.MolsToGridImage(smarts_and_mols, highlightAtomLists=matches, legends=legends)
if verbose:
return drawing, mcs_smarts, mcs_mol, mols
else:
return drawing
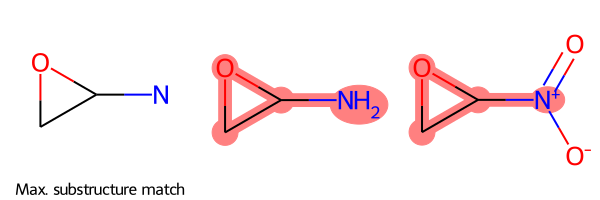
Minimal Example
All you have to provide to SmilesMCStoGridImage is a list of SMILES strings, and it will return a grid image:
SmilesMCStoGridImage(["NC1OC1", "C1OC1[N+](=O)[O-]"])
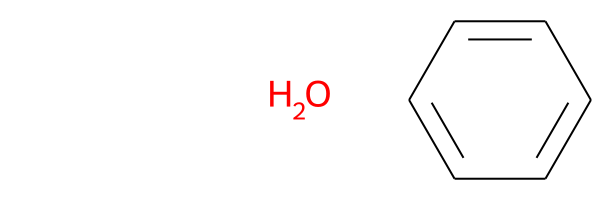
If there is no common substructure, the first cell in the grid will be empty (because there is no SMARTS match), and the molecules will be displayed without any highlighting:
SmilesMCStoGridImage(["O", "c1ccccc1"])
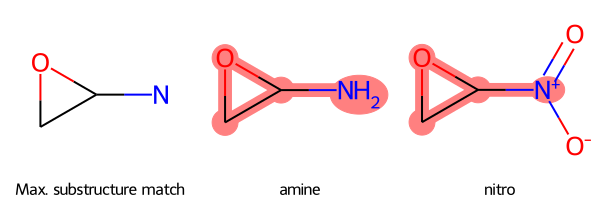
Label Molecules
If you want to label the molecules in the grid image, provide a dictionary of molecules where each
- key is the SMILES string for that molecule
- value is the legend for that molecule, for example its name or a description
SmilesMCStoGridImage({"NC1OC1": "amine", "C1OC1[N+](=O)[O-]": "nitro"})
Get Additional Data
If you want SmilesMCStoGridImage to return not just the grid image, but also the substructure match and molecule, plus the molecules for the SMILES strings, set verbose=True:
drawing, mcs_smarts, mcs_mol, mols = SmilesMCStoGridImage({"NC1OC1": "amine", "C1OC1[N+](=O)[O-]": "nitro"}, verbose=True)
You then must explicitly call the image to draw it:
drawing
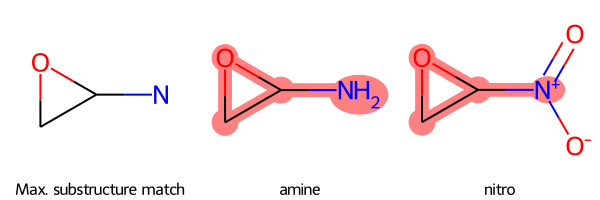
mcs_smarts is the SMARTS string for the maximum common substructure (MCS):
mcs_smarts
'[#7]-[#6]1-[#8]-[#6]-1'
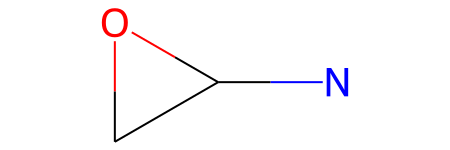
mcs_mol is the molecular representation of that MCS:
mcs_mol
mols is the list of RDKit molecules:
mols
[<rdkit.Chem.rdchem.Mol at 0x7f78a0bc0880>,
<rdkit.Chem.rdchem.Mol at 0x7f78b1215ca0>]
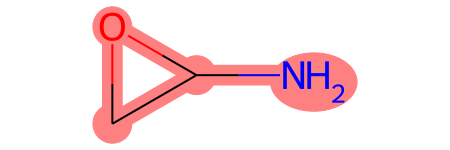
You can plot each molecule, with the MCS highlighted, by indexing the molecule in mols:
mols[0]
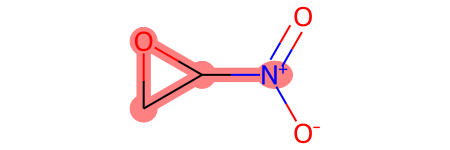
mols[1]
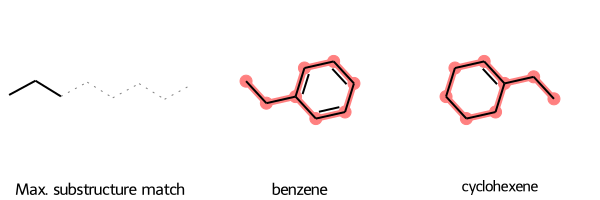
Caveat About Aligning Maximum Common Substructure
The SMARTS substructure match may not match the form of the molecule. For example, if you input two structures containing six-membered carbon rings, the SMARTS substructure match includes a linear chain of six carbons. So if you align the molecules to that substructure, you get some odd-looking “rings”:
SmilesMCStoGridImage({"CCc1ccccc1": "benzene", "C1CCCC=C1CC": "cyclohexene"})

To address this case, in SmilesMCStoGridImage you can set align_substructure=False (its default value is True), with the disadvantage that the molecules may not be aligned:
SmilesMCStoGridImage({"CCc1ccccc1": "benzene", "C1CCCC=C1CC": "cyclohexene"}, align_substructure=False)
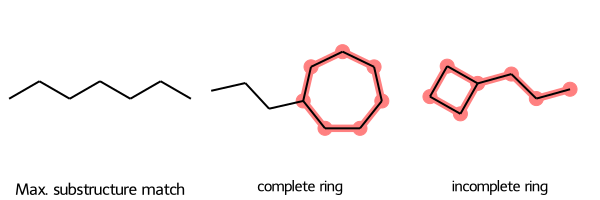
Pass Arguments to FindMCS
SmilesMCStoGridImage will pass any keyword arguments of rdFMCS.FindMCS to that function. For example, by default, rdFMCS.FindMCS will match a pattern of atoms even if they are in a complete ring in one molecule, and not in another:
SmilesMCStoGridImage({"C1CCCCCC1CCC": "complete ring", "C1CCC1CCC": "incomplete ring"}, align_substructure=False)
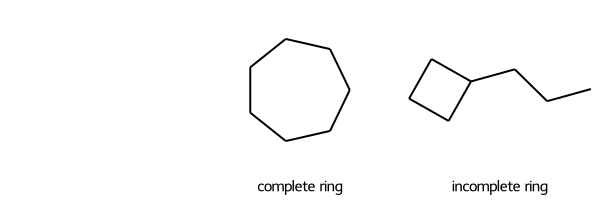
rdFMCS.FindMCS lets you set the flag completeRingsOnly=True to avoid matching these two molecules. You can call SmilesMCStoGridImage with completeRingsOnly=True to pass that flag to rdFMCS.FindMCS so that the two molecules won’t produce a match:
SmilesMCStoGridImage({"C1CCCCCC1": "complete ring", "C1CCC1CCC": "incomplete ring"}, align_substructure=False, completeRingsOnly=True)
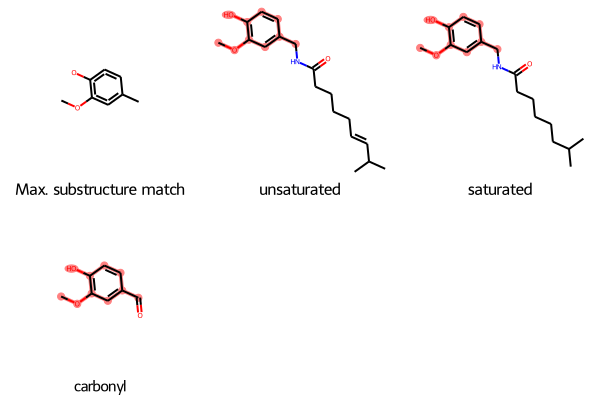
Example With Larger Molecules
As a final example, here’s a case with larger molecules:
SmilesMCStoGridImage({"O=C(NCc1cc(OC)c(O)cc1)CCCC/C=C/C(C)C": "unsaturated", "CC(C)CCCCCC(=O)NCC1=CC(=C(C=C1)O)OC": "saturated", "c1(C=O)cc(OC)c(O)cc1": "carbonyl"})