Molecular Formula Generation
In cheminformatics, the typical way of representing a molecule is with a SMILES string such as CCO for ethanol. A SMILES string can be converted into a molecular graph, which can be used to determine molecular structure and related properties. However, there are still cases where the molecular formula such as C2H6O is useful.
For example, the molecular formula is sufficient to determine the molecular mass, to calculate the predicted results from an elemental analysis, to get a sense for the elemental composition of a given molecule, or for balancing chemical equations. One of the best-known chemical equations describes photosynthesis:

I’m working on a blog post where I need to calculate the molecular formula including isotopes. I was unable to find a package that produced molecular formulas including isotopes from SMILES strings, so I wrote a function to do so. (I tried chemparse and did not succeed.)
Open this notebook in Google Colab so you can run it without installing anything on your computer
%%capture
%!pip install rdkit
%!pip install black[jupyter]
from collections import defaultdict
from IPython.display import display, Markdown
from rdkit import Chem
Setting up the molecule
Because RDKit excludes hydrogen atoms by default, but we want to include hydrogens in our molecular formula, we tell RDKit to add hydrogens.
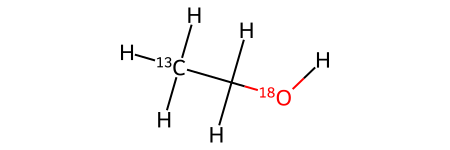
# Ethanol with isotopes
sml = "[13CH3]C[18OH]"
mol = Chem.AddHs(Chem.MolFromSmiles(sml))
mol
Composition function
The RDKit does not include a composition function to give the number of each element (including isotopes if desired) in a molecule, so we make one here. It’s based on an algorithm suggested by @IchiruTake.
def composition(
molecule: Chem.Mol,
isotopes: bool = False,
) -> defaultdict:
"""Get the composition of an RDKit molecule:
Atomic counts, including hydrogen atoms, and isotopes if requested.
For example, ethanol (SMILES [13C](H)(H)(H)CO, formula C2H6O) returns:
if isotopes = False (default): {'C': 2, 'O': 1, 'H': 6}.
if isotopes = True: {'C': {13: 1, 0: 1}, 'O': {0: 1}, 'H': {0: 6}}.
:param molecule: The RDKit molecule to analyze
:param isotopes: Whether to include the isotope of each atom
:returns: if isotopes = False (default): a dictionary of element:count entries;
if isotopes = True: a nested dictionary of element:isotope:count entries.
"""
# Check that there is a valid molecule
if not molecule:
return
# Add hydrogen atoms--RDKit excludes them by default
molecule = Chem.AddHs(molecule)
comp = defaultdict(lambda: 0)
# Get atom counts
for atom in molecule.GetAtoms():
element_symbol = atom.GetSymbol()
# If isotopes not requested, simply count the number of atoms of each element
if not isotopes:
comp[element_symbol] += 1
# If isotopes requested, count the number of each isotope of each element
else:
isotope = atom.GetIsotope()
try:
comp[element_symbol][isotope] += 1
except:
comp[element_symbol] = defaultdict(lambda: 0)
comp[element_symbol][isotope] += 1
return comp
With isotopes=False, we get the count of each element:
composition(mol, isotopes=False)
defaultdict(<function __main__.composition.<locals>.<lambda>()>,
{'C': 2, 'O': 1, 'H': 6})
With isotopes=True, the first layer (dictionary) is chemical elements, and the second layer (nested dictionary) is the isotope and the count:
composition(mol, isotopes=True)
defaultdict(<function __main__.composition.<locals>.<lambda>()>,
{'C': defaultdict(<function __main__.composition.<locals>.<lambda>()>,
{13: 1, 0: 1}),
'O': defaultdict(<function __main__.composition.<locals>.<lambda>()>,
{18: 1}),
'H': defaultdict(<function __main__.composition.<locals>.<lambda>()>,
{0: 6})})
Molecular formula generation
Now that we have the count of each element (with isotopes if desired) in the molecule, we can build the molecular formula. We need a formatting language to render superscripts for isotopes, and subscripts for counts. Our mol_to_formatted_formula() provides a dictionary with two formatting options:
- Markdown, which is commonly converted to HTML. Here the only formatting is HTML tags (for superscripts and subscripts), so the output is valid HTML as well.
- LaTeX, which is commonly used for technical papers.
A couple notes about LaTeX formatting:
- LaTeX overlaps a subscript and a superscript that immediately followed it, so in the code below we add a small amount of horizontal space using an empty group
{}. - The C-style string substitution using
%throughout this post is necessary instead of f-string{variable}or{}.format(variable)because- the braces in those last two interfere with the LaTeX formatting, or
- produce incorrect results when Markdown is converted to HTML when this post is put online.
def mol_to_formatted_formula(
mol: Chem.Mol,
isotopes: bool = False,
) -> dict[str, str]:
"""Convert an RDKit molecule to a formatted formula, in Markdown and LaTeX
:param mol: RDKit molecule
:param isotopes: Whether to consider isotopes
:returns: a dictionary of format:string pairs, e.g. {"markdown":"markdown_string", "latex":"latex_string"}
"""
if mol is None:
return "Invalid molecule"
comp = composition(mol, isotopes)
formula = {"markdown": "", "latex": ""}
if isotopes:
isotopes_dict = defaultdict(lambda: defaultdict(str))
subscripts = defaultdict(lambda: defaultdict(int))
superscripts = defaultdict(list)
for element, counts in comp.items():
for isotope, count in counts.items():
if count > 1:
subscripts[element][isotope] = count
if isotope != 0:
superscripts[element].append(isotope)
isotopes_dict[element][isotope] = 1
# Sort the element's isotopes from lowest to highest
superscripts[element].sort()
last_item_is_subscript = False
sorted_element_keys = sorted(isotopes_dict.keys())
for element in sorted_element_keys:
isotope_count_pairs = isotopes_dict[element]
# Sort the element's isotopes from lowest to highest
sorted_isotope_keys = sorted(isotope_count_pairs.keys())
for isotope in sorted_isotope_keys:
if element in superscripts:
if isotope in superscripts[element]:
formula["markdown"] += f"<sup>{isotope}</sup>"
# If superscript immediately follows subscript,
# add a small amount of horizontal space using an empty group {}
# to prevent them from vertically overlapping
if last_item_is_subscript:
formula["latex"] += "{}"
formula["latex"] += "^{ %d}" % isotope
last_item_is_subscript = False
formula["markdown"] += element
formula["latex"] += element
last_item_is_subscript = False
if element in subscripts:
if isotope in subscripts[element]:
isotope_count = subscripts[element][isotope]
formula["markdown"] += f"<sub>{isotope_count}</sub>"
formula["latex"] += "_{ %d}" % isotope_count
last_item_is_subscript = True
# Add beginning and ending dollar signs to LaTeX formula
formula["latex"] = "$" + formula["latex"] + "$"
else:
# Handling the case when isotopes is False
sorted_element_keys = sorted(comp.keys())
for element in sorted_element_keys:
count = comp[element]
formula["markdown"] += element
formula["latex"] += element
if count > 1:
formula["markdown"] += f"<sub>{count}</sub>"
formula["latex"] += "_{ %d}" % count
formula["latex"] = "$" + formula["latex"] + "$"
return formula
Here’s the Markdown for our isotopic ethanol molecule:
isotope_formula_markdown = mol_to_formatted_formula(mol, isotopes=True)["markdown"]
isotope_formula_markdown
'C<sup>13</sup>CH<sub>6</sub><sup>18</sup>O'
and we display it as Markdown using Markdown():
Markdown(isotope_formula_markdown)
C13CH618O
Here’s the LaTeX version:
isotope_formula_latex = mol_to_formatted_formula(mol, isotopes=True)["latex"]
isotope_formula_latex
'$C^{ 13}CH_{ 6}{}^{ 18}O$'
which can also be displayed using Markdown():
Markdown(isotope_formula_latex)
$C^{ 13}CH_{ 6}{}^{ 18}O$
As far as the order of elements in a chemical formula, mol_to_formatted_formula() simply alphabetizes them. So the elements some formulas may not appear in the typical order. Within an element, mol_to_formatted_formula() gives the isotopes in increasing order, with the unspecified isotope first.
To go directly from a SMILES string to a formula, we can use this utility function smiles_to_formatted_formula():
def smiles_to_formatted_formula(smiles: str, isotopes: bool = False) -> dict[str,str]:
"""Convert a SMILES string to a formatted formula, in Markdown and LaTeX
:param smiles: SMILES string
:param isotopes: Whether to consider isotopes
:returns: a dictionary of format:string pairs, e.g. {"markdown":"markdown_string", "latex":"latex_string"}
"""
mol = Chem.MolFromSmiles(smiles)
if mol is not None:
return mol_to_formatted_formula(mol, isotopes=isotopes)
isotope_formula_latex_from_smiles = smiles_to_formatted_formula(sml, isotopes=True)[
"latex"
]
Markdown(isotope_formula_latex_from_smiles)
$C^{ 13}CH_{ 6}{}^{ 18}O$
Improved formatting using LaTeX
LaTeX italicizes letters by default, so we can use LaTeX \mathrm to make them non-italicized.
def markdown_formula(latex: str) -> str:
"""Make a LaTeX molecular formula non-italicized by removing math formatting
:param latex: the molecular formula
:returns: the non-italicized molecular formula
"""
latex_markdown = r"$\mathrm{ %s}$" % (latex.strip("$"))
return latex_markdown
Here’s the non-italicized result:
Markdown(markdown_formula(isotope_formula_latex))
$\mathrm{ C^{ 13}CH_{ 6}{}^{ 18}O}$
As a further utility, we can immediately display the result as Markdown by incorporating that function:
def display_markdown_formula(latex: str) -> str:
"""Display a LaTeX molecular formula, non-italicized
:param latex: the molecular formula
:returns: Markdown display of the non-italicized molecular formula
"""
latex_markdown = r"$\mathrm{ %s}$" % (latex.strip("$"))
return Markdown(latex_markdown)
display_markdown_formula(isotope_formula_latex)
$\mathrm{ C^{ 13}CH_{ 6}{}^{ 18}O}$
Conclusion
Now that we have a way to calculate molecular formulas, and two formats to display them in, the next blog post will give applications of each format.
Postscripts
I later learned from Wim that there is an RDKit function to create a non-formatted molecular formula, CalcMolFormula. One could probably parse its output to produce formatted formulas, though I find that parsing strings is finnicky, and it would probably be about the same amount of effort as the approach above.
from rdkit.Chem.rdMolDescriptors import CalcMolFormula
print(CalcMolFormula(mol, separateIsotopes=True))
C[13C]H6[18O]
Here’s how to use LaTeX to create the photosynthesis chemical equation shown at the top of the blog post.
photosynthesis_smls = {
"Water": "O",
"Oxygen": "O=O",
"Carbon Dioxide": "O=C=O",
"Glucose": "OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O",
}
mols = {name: Chem.MolFromSmiles(sml) for name, sml in photosynthesis_smls.items()}
formulas = {
name: markdown_formula(mol_to_formatted_formula(mol)["latex"])
for name, mol in mols.items()
}
photosynthesis = (
"$"
+ "6"
+ formulas["Carbon Dioxide"].strip("$")
+ "+ 6"
+ formulas["Water"].strip("$")
+ "→"
+ formulas["Glucose"].strip("$")
+ "+ 6"
+ formulas["Oxygen"].strip("$")
+ "$"
)
display(Markdown(photosynthesis))
$6\mathrm{ CO_{ 2}}+ 6\mathrm{ H_{ 2}O}→\mathrm{ C_{ 6}H_{ 12}O_{ 6}}+ 6\mathrm{ O_{ 2}}$